4. Alignment of reads to a reference genome¶
The metagenomic screenig of the shotgun library detected reads assigned to Yersinia pestis. The following step is to ascertain that these molecules are authentic.
You can do that by mapping your pre-processed fastq files (merged and trimmed) to the Yersinia pestis CO92 strain reference sequence, available in the RefSeq NCBI database.
Here, in order to obtain an optimal coverage for the subsequent variant call, we will run the alignment on a different fastq file that we prepared to simulate an enriched library.
4.1. Preparation of the reference sequence¶
4.1.1. Index the reference sequence with bwa¶
To align the reads to the reference sequence we will use the program BWA, in particular the BWA aln algorithm. BWA first needs to construct the FM-index for the reference genome, with the command BWA index. FM-indexing in Burrows-Wheeler transform is used to efficiently find the number of occurrences of a pattern within a compressed text, as well as locate the position of each occurrence. It is an essential step for querying the DNA reads to the reference sequence. This command generates five files with different extensions: amb, ann, bwt, pac, sa.
bwa index -a is reference.fasta
Note
The option -a indicates the algorithm to use for constructing the index. For genomes smaller than < 2 Gb use the is algorithm. For larger genomes (>2 Gb), use the bwtsw algorithm.
4.1.2. Create a reference dictionary¶
A dictionary file (dict) is necessary to run later in the pipeline GATK RealignerTargetCreator. A sequence dictionary contains the sequence name, sequence length, genome assembly identifier, and other information about the sequences. To create the dict file we use Picard.
picard CreateSequenceDictionary R= referece.fasta O= reference.dict
Note
In our server environment we can call Picard just by typing the program name. In other environments (including your laptop) you may have to call Picard by providing the full path to the java file jar of the program:
java -jar /path/to/picard.jar CreateSequenceDictionary R= referece.fasta O= ref.dict
4.1.3. Index the reference sequence with Samtools¶
The reference sequence has to be indexed in order to run later in the pipeline GATK IndelRealigner. To do that, we will use Samtools, in particular the tool samtools faidx, which enables efficient access to arbitrary regions within the reference sequence. The index file typically has the same filename as the corresponding reference sequece, with the extension fai appended.
samtools faidx reference.fasta
4.2. Alignment of the reads to the reference sequence¶
4.2.1. Alignment of pre-processed reads to the reference genome with BWA aln¶
To align the reads to the reference genome we will use BWA aln, which supports an end-to-end alignment of reads to the reference sequence. The alternative algorithm, BWA mem supports also local (portion of the reads) and chimeric alignments (resulting in a larger number of mapped reads than BWA aln). BWA aln is more suitable for aliging short reads, like expected for ancient DNA samples. The following comand will generate a sai file.
bwa aln reference.fasta filename.fastq.gz -n 0.1 -l 1000 > filename.sai
Some of the options available in BWA aln:
| Option | Function |
|---|---|
| -n number | Maximum edit distance if the value is an integer. If the value is float the edit distance is automatically chosen for different read lengths (default=0.04) |
| -l integer | Seed length. If the value is larger than the query sequence, seeding will be disabled. |
| -o integer | Maximum number of gap opens. For aDNA, tolerating more gaps helps mapping more reads (default=1). |
Note
- Due to the particular damaged nature of ancient DNA molecules, carrying deaminations at the molecules ends, we deactivate the
seed-lengthoption by giving it a high value (e.g.-l 1000). - Here we are aligning reads to a bacterial reference genome. To reduce the impact of spurious alignemnts due to presence bacterial species closely related to the one that we are investigating, we will adopt stringent conditions by decreasing the
maximum edit distanceoption (-n 0.1). For alignment of DNA reads to the human reference sequence, less stringent conditions can be used (-n 0.01and-o 2).
Once obtained the sai file, we align the reads (fastq file) to the reference (fasta file) using BWA samse, to generate the alignment file sam.
bwa samse reference.fasta filename.sai filename.fastq.gz -f filename.sam
4.2.2. Converting sam file to bam file¶
For the downstream analyses we will work with the binary (more compact) version of the sam file, called bam. To convert the sam file in bam we will use Samtools view.
samtools view -Sb filename.sam > filename.bam
Note
The conversion from sam to bam can be piped (|) in one command right after the alignment step:
bwa samse reference.fasta filename.sai filename.fastq.gz | samtools view -Sb - > filename.bam
To view the content of a sam file we can just use standard commands like head, tail, less, while to view the content of a bam file (binary format of sam) we have to use Samtools view:
samtools view filename.bam
You may want to display on the screen one read/line (scrolling with the spacebar):
samtools view filename.bam | less -S
while to display just the header of the bam file:
samtools view -H filename.bam
4.2.3. Sorting and indexing the bam file¶
To go on with the analysis, we have to sort the reads aligned in the bam file by leftmost coordinates (or by read name when the option -n is used) with Samtools sort. The option -o is used to provide an output file name:
samtools sort filename.bam -o filename.sort.bam
The sorted bam files are then indexed with Samtools index. Indexes allow other programs to retrieve specific parts of the bam file without reading through each sequence. The following command generates a bai file, a companion file of the bam which contains the indexes:
samtools index filename.sort.bam
4.2.4. Adding Read Group tags and indexing bam files¶
A number of predefined tags may be appropriately assigned to specific set of reads in order to distinguish samples, libraries and other technical features. To do that we will use Picard. You may want to use RGLB (library ID) and RGSM (sample ID) tags at your own convenience based on the experimental design. Remember to call Picard from the path of the jar file.
picard AddOrReplaceReadGroups INPUT= filename.sort.bam OUTPUT= filename.RG.bam RGID=rg_id RGLB=lib_id RGPL=platform RGPU=plat_unit RGSM=sam_id VALIDATION_STRINGENCY=LENIENT
Note
- In some instances, Picard may stop running and return error messages due to conflicts with
samspecifications produced byBWA(e.g. “MAPQ should be 0 for unmapped reads”). To suppress this error and allow Picard to continue, we pass theVALIDATION_STRINGENCY=LENIENToptions (default isSTRICT). - Read Groups may be also added during the alignment with
BWAusing the option-R.
Once added the Read Group tags, we index again the bam file:
samtools index filename.RG.bam
4.2.5. Marking and removing duplicates¶
Amplification through PCR of genomic libraries leads to duplication formation, hence reads originating from a single fragment of DNA. The MarkDuplicates tool of Picard marks the reads as duplicates when the 5’-end positions of both reads and read-pairs match. A metric file with various statistics is created, and reads are removed from the bam file by using the REMOVE_DUPLICATES=True option (the default option is False, which simply ‘marks’ duplicate reads keep them in the bam file).
picard MarkDuplicates I= filename.RG.bam O= filename.DR.bam M=output_metrics.txt REMOVE_DUPLICATES=True VALIDATION_STRINGENCY=LENIENT &> logFile.log
Once removed the duplicates, we index again the bam file:
samtools index filename.DR.bam
4.2.6. Local realignment of reads¶
The presence of insertions or deletions (indels) in the genome may be responsible of misalignments and bases mismatches that are easily mistaken as SNPs. For this reason, we locally realign reads to minimize the number of mispatches around the indels. The realignment process is done in two steps using two different tools of GATK called with the -T option. We first detect the intervals which need to be realigned with the GATK RealignerTargetCreator, and save the list of these intevals in a file that we name target.intervals:
gatk -T RealignerTargetCreator -R reference.fasta -I filename.DR.bam -o target.intervals
Note
Like Picard, in some server environment you can call GATK just by typing the program name. In other environments (also in this server) you have to call GATK by providing the full path to the java jar file. Here, the absolute path to the file is ~/Share/Paleogenomics/programs/GenomeAnalysisTK.jar:
java -jar ~/Share/Paleogenomics/programs/GenomeAnalysisTK.jar -T RealignerTargetCreator -h
Warning
In version 4 of GATK the indel realigment tools have been retired from the best practices (they are unnecessary if you are using an assembly based caller like Mutect2 or HaplotypeCaller). To use the indel realignment tools make sure to install version 3 of GATK.
Then, we realign the reads over the intervals listed in the target.intervals file with the option -targetIntervals of the tool IndelRealigner in GATK:
java -jar ~/Share/Paleogenomics/programs/GenomeAnalysisTK.jar -T IndelRealigner -R reference.fasta -I filename.RG.DR.bam -targetIntervals target.intervals -o filename.final.bam --filter_bases_not_stored
Note
- If you want, you can redirect the standard output of the command into a
logfile by typing at the end of the command&> logFile.log - The option
--filter_bases_not_storedis used to filter out reads with no stored bases (i.e. with * where the sequence should be), instead of failing with an error
The final bam file has to be sorted and indexed as previously done:
samtools sort filename.final.bam -o filename.final.sort.bam
samtools index filename.final.sort.bam
4.2.7. Generate flagstat file¶
We can generate a file with useful information about our alignment with Samtools flagstat. This file is a final summary report of the bitwise FLAG fields assigned to the reads in the sam file.
samtools flagstat filename.final.sort.bam > flagstat_filename.txt
Note
You could generate a flagstat file for the two
bamfiles before and after refinement and see the differences.You can decode each
FLAGfield assigned to a read on the Broad Institute website.
4.2.8. Visualization of reads alignment¶
Once generated the final bam file, you can compare the bam files before and after the refinement and polishing process (duplicates removal, realignment around indels and sorting). To do so, we will use the program IGV, in which we will first load the reference fasta file from Genomes –> Load genome from file and then we will add one (or more) bam files with File –> Load from file:
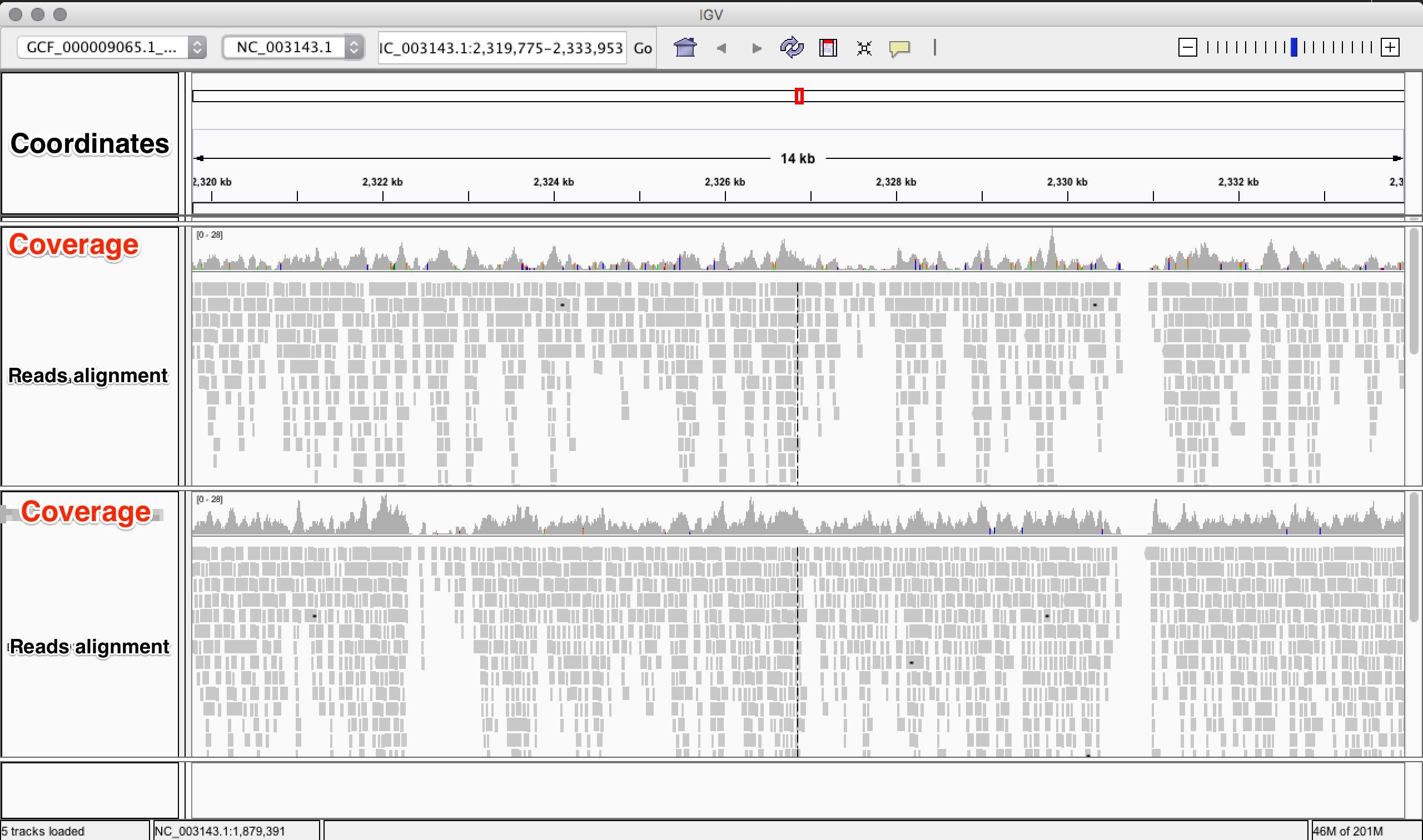
4.3. Create mapping summary reports¶
We will use Qualimap to create summary reports from the generated bam files. As mentioned in the website, Qualimap examines sequencing alignment data in sam/bam files according to the features of the mapped reads and provides an overall view of the data that helps to detect biases in the sequencing and/or mapping of the data and eases decision-making for further analysis.
qualimap bamqc -c -bam input.bam
Here are some screenshots of the outputs:
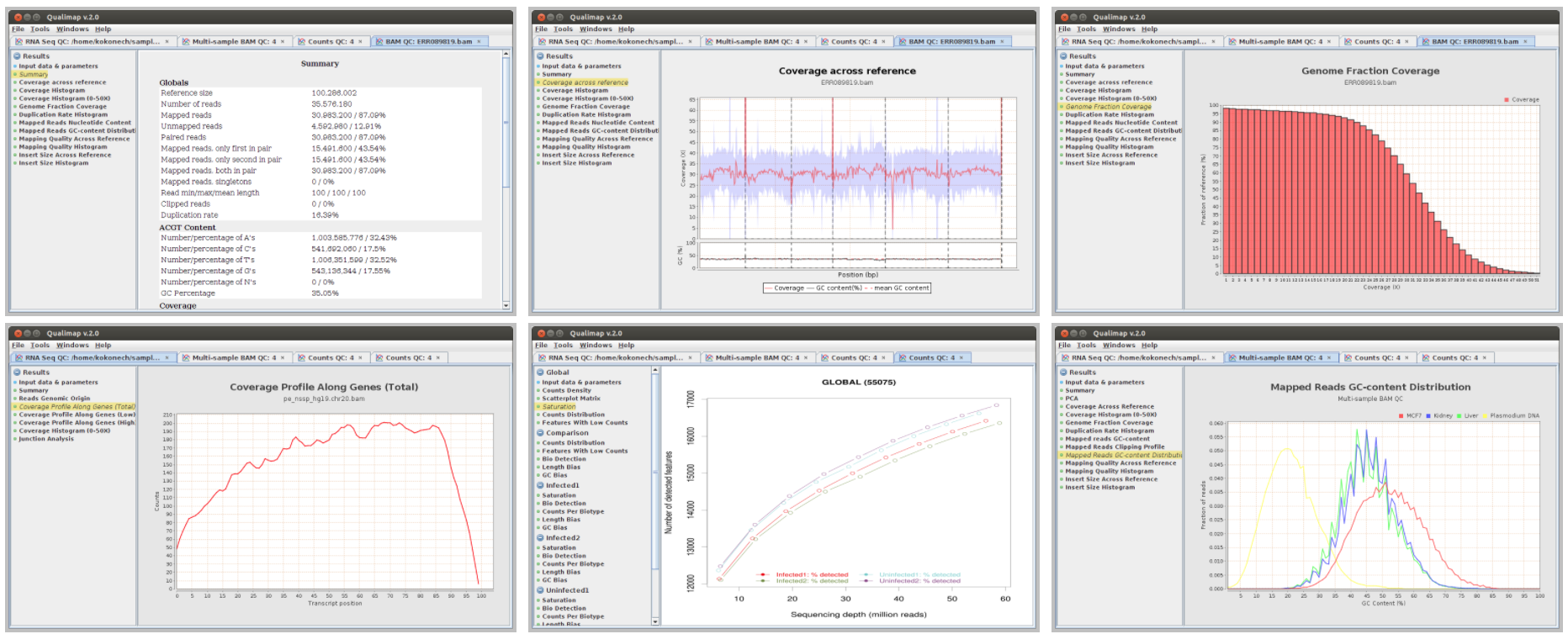
At this stage we have created different type of summary report using FastQC and Qualimap. To create a unique summary that integrate and compare all the generated reports, we will use MultiQC. If all the reports are in the same directory and its sub-directories, you can run MultiQC as follows:
multiqc .
A list of programs that generate output files recognized by MultiQC are availble here: https://github.com/ewels/MultiQC
Multiqc will create a summary report in html format that will let you compare all the summary reports for each of your samples:
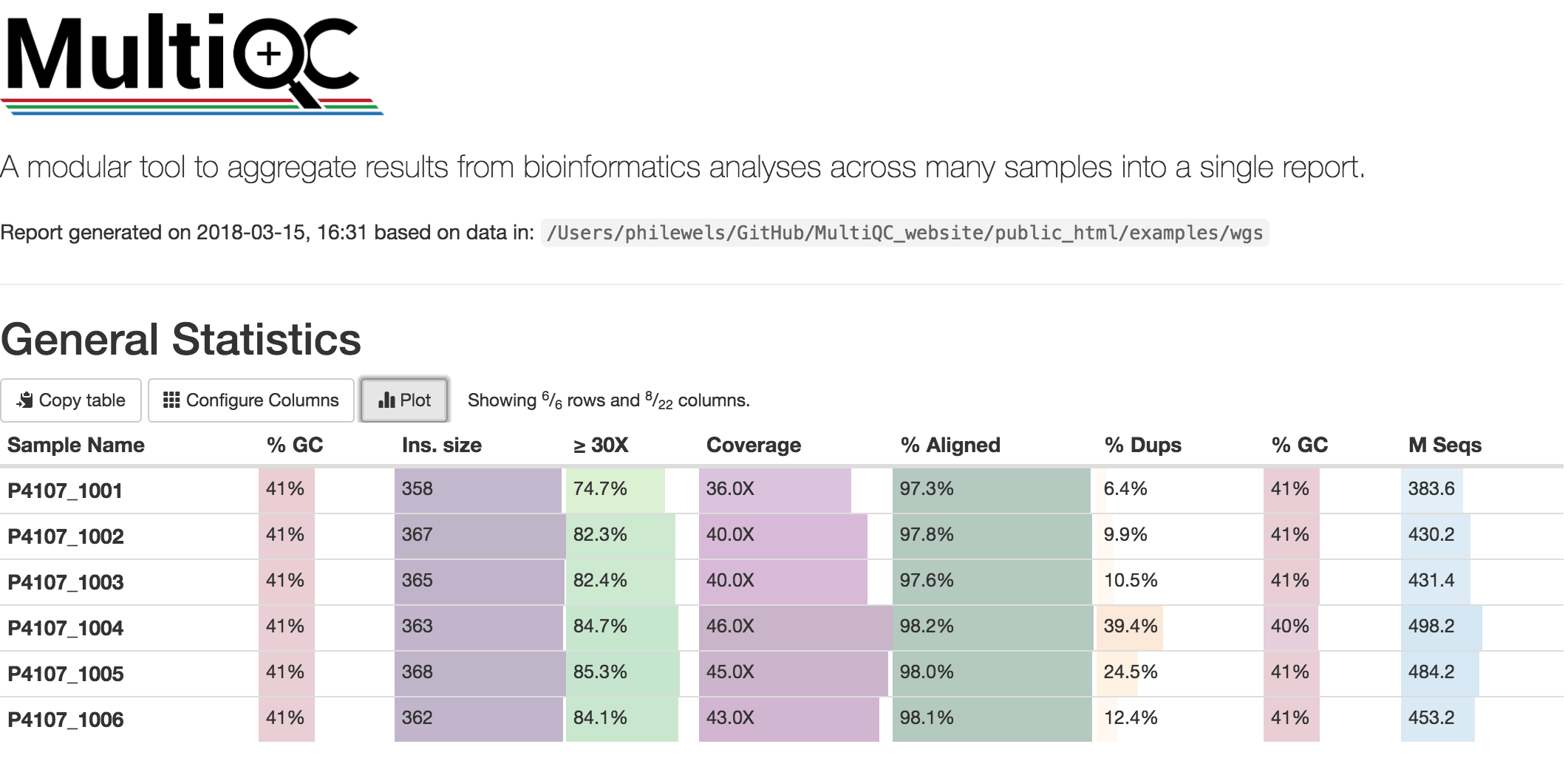
4.4. Damage analysis and quality rescaling of the BAM file¶
To authenticate our analysis we will assess the post-mortem damage of the reads aligned to the reference sequence. We can track the post-portem damage accumulated by DNA molecules in the form of fragmentation due to depurination and cytosine deamination, which generates the typical pattern of C->T and G->A variation at the 5’- and 3’-end of the DNA molecules. To assess the post-mortem damage patterns in our bam file we will use mapDamage, which analyses the size distribution of the reads and the base composition of the genomic regions located up- and downstream of each read, generating various plots and summary tables. To start the analysis we need the final bam and the reference sequence:
mapDamage -i filename.final.sort.bam -r reference.fasta
mapDamage creates a new folder where the output files are created. One of these files, is named Fragmisincorporation_plot.pdf which contains the following plots:
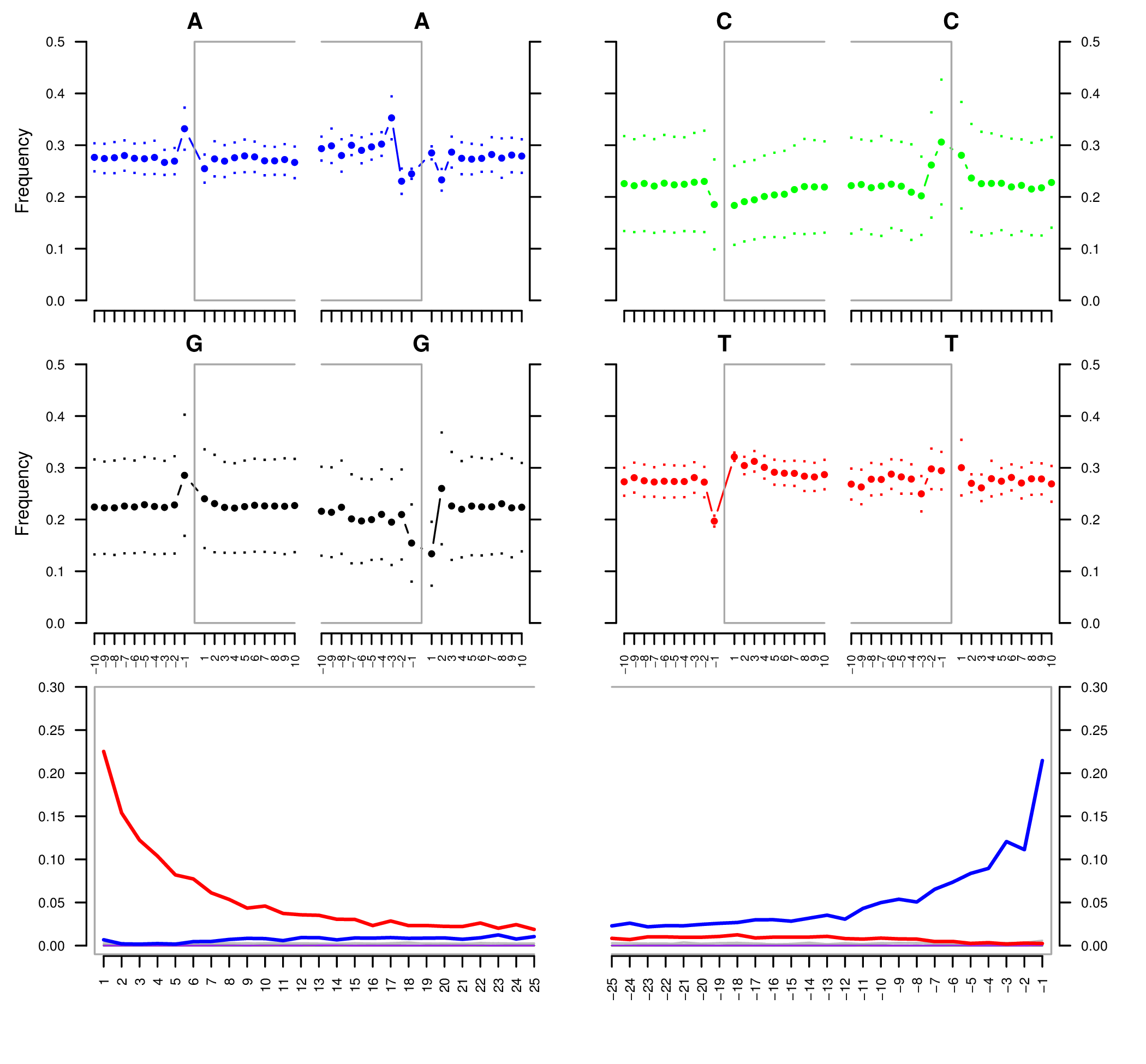
If DNA damage is detected, we can run mapDamage again using the --rescale-only option and providing the path to the results folder that has been created by the program (option -d). This command will downscale the quality scores at positions likely affected by deamination according to their initial quality values, position in reads and damage patterns.
A new rescaled bam file is then generated.
mapDamage -i filename.final.sort.bam -r reference.fasta --rescale-only -d results_folder
You can also rescale the bam file directly in the first command with the option --rescale:
mapDamage -i filename.final.sort.bam -r reference.fasta --rescale
Note
Another useful tool for estimating post-mortem damage (PMD) is PMDTools. This program uses a model incorporating PMD, base quality scores and biological polymorphism to assign a PMD score to the reads. PMD > 0 indicates support for the sequence being genuinely ancient. PMDTools filters the damaged reads (based on the selected score) in a separate bam file which can be used for downstream analyses (e.g. variant call).
The rescaled bam file has to be indexed, as usual.
samtools index filename.final.sort.rescaled.bam
4.5. Edit Distance¶
The edit distance defines the number of nucleotide changes that have to be made to one read sequence for it to be identical to the reference sequence.
To be more confident about the quality and authenticity of your sequencing data, you need to align your reads againt your reference sequence and the genome of a closely related species.
Here we will align our fastq file against the Yersinia pseudotuberculosis genome, following all the steps from 4.1 to 4.4.
The edit distance must be lower when aligning the reads to the reference sequence compared to the closely related species.

To calculate the edit distance we will use BAMStats, a tool for summarising Next Generation Sequencing alignments. The commands to generate summary-charts, including the edit distance is:
BAMStats -i filename.rescaled.bam -v html -d -q -o outfile.html
| Option | Function |
|---|---|
| -i filename | SAM or BAM input file (must be sorted). |
| -v html/simple | View option for output format (currently accepts ‘simple’ or ‘html’; default, simple). |
| -d | If selected, edit distance statistics will also be displayed as a separate table (optional). |
| -q | If selected, mapping quality (MAPQ) statistics will also be displayed as a separate table (optional). |