5. Variant calling and visualization¶
Once the reads are aligned and the data authenticated through post-mortem damage analysis, we can analyse the variant positions in the samples against the reference sequence.
5.1. Variants calling¶
We will use two common tools for variants calling: Samtools, in particular samtools mpileup, in combination with bcftools call of the program BCFtools.
samtools mpileup -B -ugf reference.fasta filename.final.sort.rescaled.bam | bcftools call -vmO z - > filename.vcf.gz
| Samtools mpileup options | Function |
|---|---|
| -B, –no-BAQ | BAQ is the Phred-scaled probability of a read base being misaligned. Applying this option greatly helps to reduce false SNPs caused by misalignments. |
| -u, –uncompressed | Generate uncompressed VCF/BCF output, which is preferred for piping. |
| -g, –BCF | Compute genotype likelihoods and output them in the binary call format (BCF). As of v1.0, this is BCF2 which is incompatible with the BCF1 format produced by previous (0.1.x) versions of samtools. |
| -f, –fasta-ref file | The faidx-indexed reference file in the FASTA format. The file can be optionally compressed by bgzip. |
| BCFtools call options | Function |
|---|---|
| -v, –variants-only | Output variant sites only. |
| -m, –multiallelic-caller | Alternative modelfor multiallelic and rare-variant calling designed to overcome known limitations in -c calling model (conflicts with -c) |
| -g, –BCF | Compute genotype likelihoods and output them in the binary call format (BCF). As of v1.0, this is BCF2 which is incompatible with the BCF1 format produced by previous (0.1.x) versions of samtools. |
| -O, –output-type b|u|z|v | Output compressed BCF (b), uncompressed BCF (u), compressed VCF (z), uncompressed VCF (v). |
The detected genetic variants will be stored in the vcf file. The genetic variants can be filtered according to some criteria using BCFtools:
bcftools filter -O z -o filename.filtered.vcf -s LOWQUAL -i'%QUAL>19' filename.vcf.gz
| BCFtools filter options | Function |
|---|---|
| -O, –output-type b|u|z|v | Output compressed BCF (b), uncompressed BCF (u), compressed VCF (z), uncompressed VCF (v). |
| -o, –output file | Output file. |
| -s, –soft-filter string|+ | Annotate FILTER column with <string> or, with +, a unique filter name generated by the program (“Filter%d”). |
| -i, –include expression | Include only sites for which expression is true. |
Note
other options can be added when using BCFtools filter:
Option Function -g, –SnpGap int Filter SNPs within int base pairs of an indel -G, –IndelGap int Filter clusters of indels separated by int or fewer base pairs allowing only one to pass
Instead of samtools mpileup and bcftools call (or in addition to) we can use gatk HaplotypeCaller:
java -jar GenomeAnalysisTK.jar -T HaplotypeCaller -R reference.fasta -I filename.final.sort.rescaled.bam -o original.vcf.gz
java -jar GenomeAnalysisTK.jar -T VariantFiltration -R reference.fasta -V filename.vcf.gz -o filename.filtered.vcf.gz --filterName 'Cov3|Qual20' --filterExpression 'DP>2||QUAL>19'
Now that you have your vcf file, you can open the file (use nano or vim in the server, or download the file in your laptop with scp and open it in a text editor) and try to search diagnostic variants (e.g. for classification). You can also visualize the variants in a specific program, as described below.
5.2. Variants visualization¶
To be able to visualize the variants in the vcf files, you can use the program IGV, which accepts multiple input files formats eg. fasta, bam, vcf and gff. After loading your bam file(s) and the corrsponding vcf file(s), you will see something likt that:
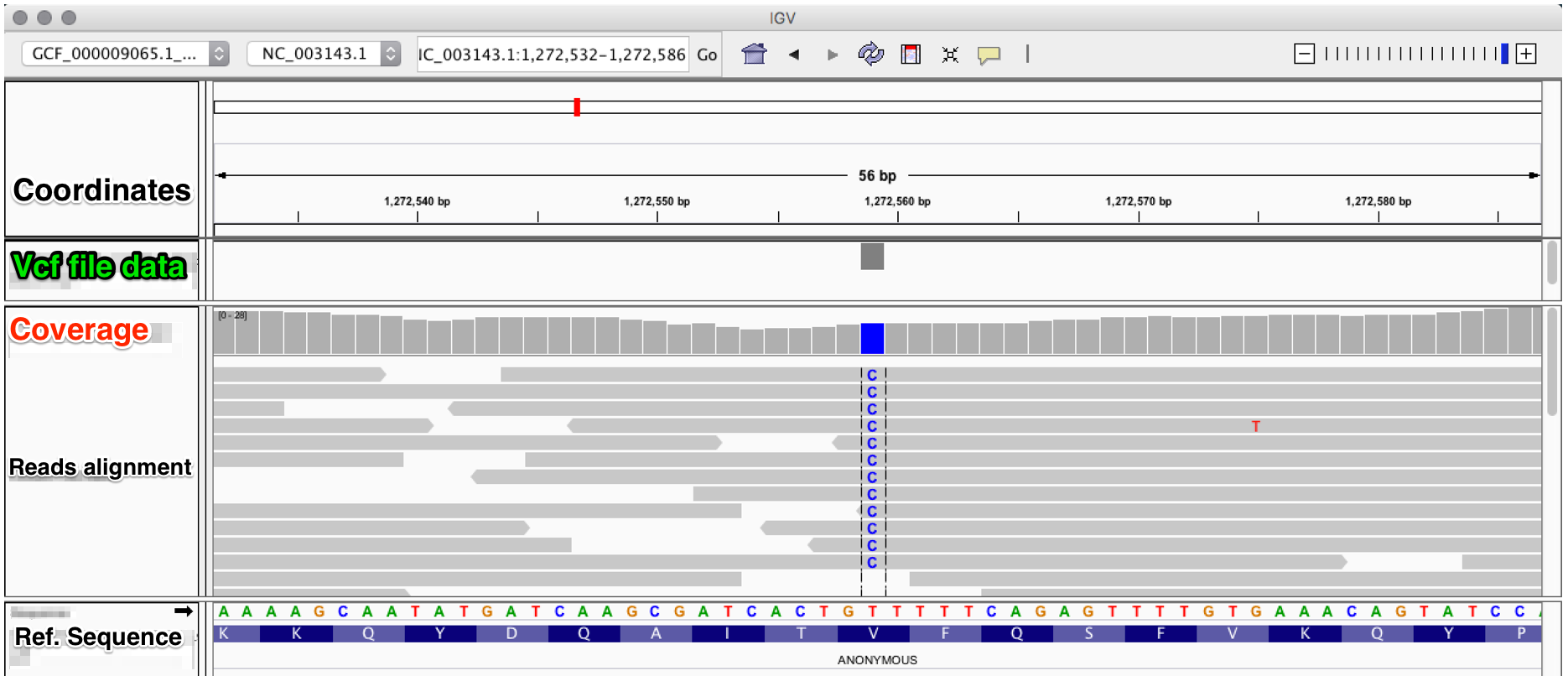
In this figure, we observe in the bam alignment file a T->C transition in the corresponding position.