3. Metagenomic screening of shotgun data¶
3.1. Kraken¶
In this hands-on session we will use Kraken to screen the metagenomic content of a DNA extract after shotgun sequencing. A Kraken database is a directory containing at least 4 files:
- database.kdb: Contains the k-mer to taxon mappings
- database.idx: Contains minimizer offset locations in database.kdb
- taxonomy/nodes.dmp: Taxonomy tree structure + ranks
- taxonomy/names.dmp: Taxonomy names
3.1.1. Minikraken¶
We will first use a pre-built 8 GB Kraken database, called Minikraken, constructed from complete dusted bacterial, archaeal, and viral genomes in RefSeq (as of October 2017).
You can download the pre-built Minikraken database from the website with wget, and extract the archive content with tar:
wget https://ccb.jhu.edu/software/kraken/dl/minikraken_20171101_8GB_dustmasked.tgz
tar -xvzf minikraken_20171101_8GB_dustmasked.tgz
Then, we can run the taxonomic assignation of the reads in our sample with the kraken command
kraken --db minikraken_20171101_8GB_dustmasked --fastq-input filename.gz --gzip-compressed --output filename.kraken
Some of the options available in Kraken:
| Option | Function |
|---|---|
| –db <string> | Path to the folder (database name) containing the database files. |
| –output <string> | Print output to filename. |
| –threads <integer> | Number of threads (only when multiple cores are used). |
| –fasta-input | Input is FASTA format. |
| –fastq-input | Input is FASTQ format. |
| –gzip-compressed | Input is gzip compressed. |
3.1.2. Create report files¶
Once the taxonomic assignation is done, from the Kraken output file we create a report of the analysis by running the kraken-report script. Note that the database used must be the same as the one used to generate the output file in the command above. The output file is a tab-delimited file with the following fields, from left to right:
- Percentage of reads covered by the clade rooted at this taxon
- Number of reads covered by the clade rooted at this taxon
- Number of reads assigned directly to this taxon
- A rank code, indicating (U)nclassified, (D)omain, (K)ingdom, (P)hylum, (C)lass, (O)rder, (F)amily, (G)enus, or (S)pecies. All other ranks are simply ‘-‘.
- NCBI taxonomy ID
- Indented scientific name
Notice that we will have to redirect the output to a file with >.
kraken-report --db Minikraken filename.kraken > filename.kraken.report
Note
We can use a for loop to make the taxonomic assignation and create the report file for multiple samples. Notice the assignation of variables filename and fname to return output files named after the sample.
for i in *.fastq
do
filename=$(basename "$i")
fname="${filename%.fastq}"
kraken --db Minikraken --threads 4 --fastq-input $i --output /${fname}.kraken
kraken-report --db Minikraken ${fname}.kraken > ${fname}.kraken.report
done
3.1.3. Visualization of data with Krona¶
Finally, we can visualize the results of the Kraken analysis with Krona, which disaplys hierarchical data (like taxonomic assignation) in multi-layerd pie charts.
The interactive charts created by Krona are in the html format and can be viewed with any web browser.
We will convert the kraken output in html format using the program ktImportTaxonomy, which parses the information relative to the query ID and the taxonomy ID.
ktImportTaxonomy -q 2 -t 3 filename.kraken -o filename.kraken.html
Some of the options available in ktImportTaxonomy:
| Option | Function |
|---|---|
| -q <integer> | Column of input files to use as query ID. |
| -t <integer> | Column of input files to use as taxonomy ID. |
| -o <string> | Output file name. |
Note
If you want to analyze multiple kraken files from various samples you view the results in one single html file running ktImportTaxonomy as follows:
ktImportTaxonomy -q 2 -t 3 filename_1.kraken filename_2.kraken ... filename_n.kraken -o all_samples.kraken.html
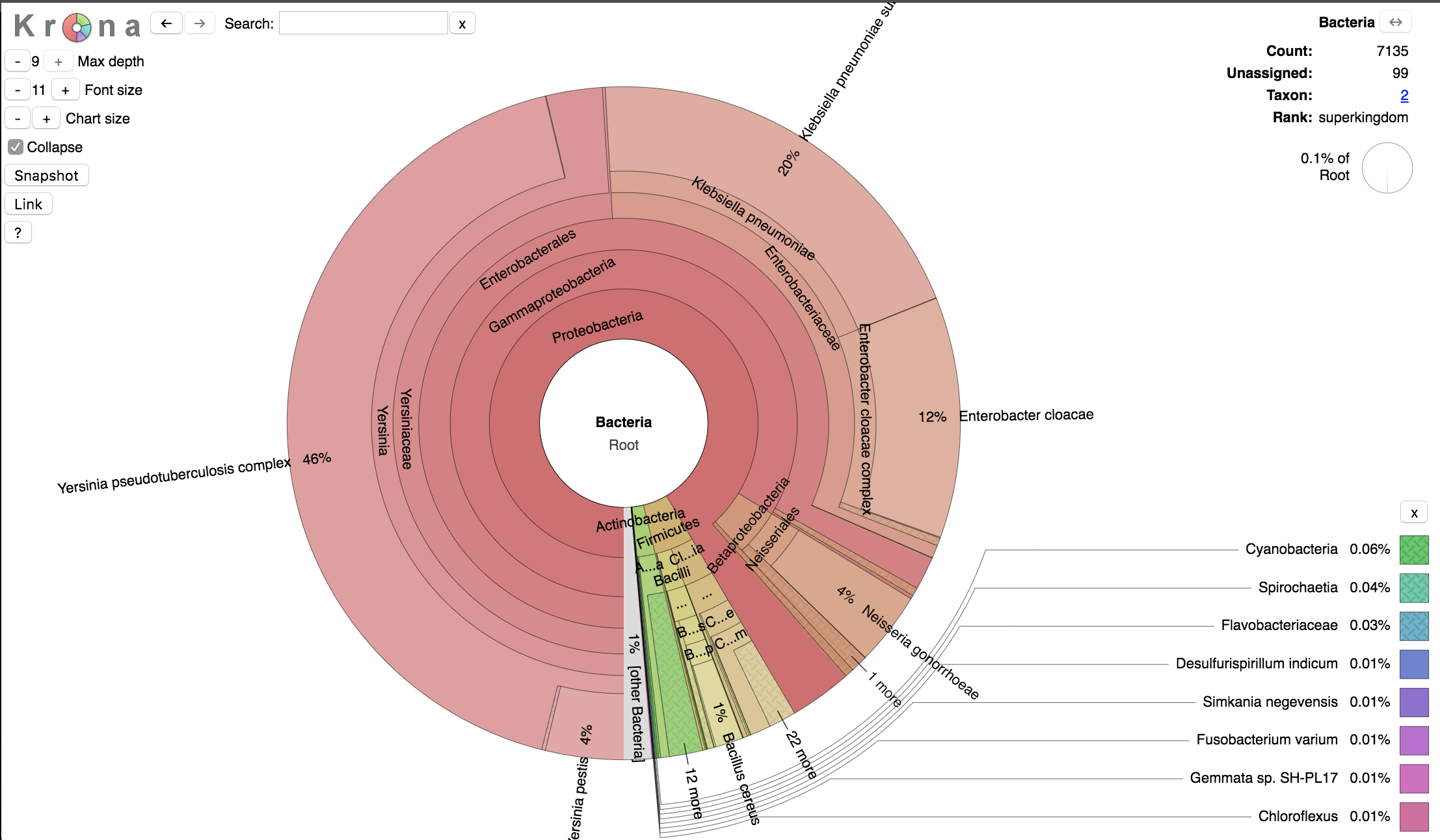
3.1.4. Building a Kraken standard database (on HPC clusters)¶
The pre-built Minikraken database is useful for a quick metagenomic screening of shotgun data. However, by building larger databases (i.e. a larger set of k-mers gathered) we may increase the sensitivity of the analysis.
One option is to build the Kraken standard database. To create this database we use the command kraken-build, which downlads the RefSeq complete genomes for the bacterial, archaeal, and viral domains, and builds the database.
kraken-build --standard --db standardkraken.folder
Note
Usage of the database will require users to keep only the
database.idx,database.kdb,taxonomy/nodes.dmpandtaxonomy/names.dmpfiles. During the database building process some intermediate file are created that may be removed afterwards with the command:kraken-build --db standardkraken.folder --clean
The downloaded RefSeq genomes require 33GB of disk space. The build process will then require approximately 450GB of additional disk space. The final
database.idx,database.kdb, andtaxonomy/files require 200 Gb of disk space, and running one sample against such database requires 175 Gb of RAM.
3.1.5. Building a Kraken custom database (on HPC clusters)¶
Building kraken custum databases is computationally intensive. You will find a ready to use database. Kraken also allows creation of customized databases, where we can choose which sequences to include and the final size of the database. For example if you do not have the computational resources to build and run analyses with a full database of bacterial genomes (or you don’t need to), you may want to build a custom database with only the genomes needed for your application.
First of all we choose a name for our database and we create a folder with that name using
mkdir. Let’s call the databaseCustomDB. This will be the name used in all the dollowing commands after the--dboption.Download NCBI taxonomy files (the sequence ID to taxon map, the taxonomic names and tree information) with
kraken-build --download-taxonomy. The taxonomy files are necessary to associate a taxon to the sequence identifier (the GI number in NCBI) of thefastasequences composing our database. For this reason we will build our database only with sequences from the NCBI RefSeq. For more information on the NCBI taxonomy visit click here. This command will create a sub-foldertaxonomy/inside our CustomDB folder:kraken-build --download-taxonomy --threads 4 --db CustomDB
Install a genomic library. RefSeq genomes in fasta file from five standard groups are made easily available in Kraken with the command kraken-build –download-library:
- bacteria : RefSeq complete bacterial genomes
- archaea : RefSeq complete archaeal genomes
- plasmid : RefSeq plasmid sequences
- viral : RefSeq complete viral genomes
- human : GRCh38 human genome
The following command will download all the RefSeq bacterial genomes (33Gb size) and create a folder
library/with a sub-folderbacteria/inside your CustomDB folder:kraken-build --download-library bacteria --threads 4 --db CustomDB
We can add any sort of RefSeq
fastasequences to the library withkraken-build --add-to-library. For example we will add to the library of bacterial genomes the RefSeq sequences of mitochodrial genomes. The sequences will be inside the sub-folderadded/.kraken-build --add-to-library mitochondrion.1.1.genomic.fna --threads 4 --db CustomDB kraken-build --add-to-library mitochondrion.2.1.genomic.fna --threads 4 --db CustomDB
Note
If you have several
fastafiles to add you can use aforloop:for i in *.fasta do kraken-build --add-to-library $i --threads 4 --db CustomDB done
When analyzing a metagenomics sample using a
Krakendatabase the primary source of false positive hits is represented by low-complexity sequences in the genomes themselves (e.g., a string of 31 or more consecutive A’s). For this reason, once gathered all the genomes that we want to use for our custom database, low-complexity regions have to be ‘dusted’. The programdustmaskerfrom Blast+ identifies low-complexity regions and soft-mask them (the corresponding sequence is turned to lower-case letters). With aforloop we run dustmasker on eachfastafile present in the library folder, and we will pipe (|) to dustmasker asedcommand to replace the low-complexity regions (lower-case) with Ns. Notice that the output is redirected (>) to a temporary file, which is afterwards renamed to replace the original filefastafile with the commandmv.for i in `find CustomDB/library \( -name '*.fna' -o -name '*.ffn' \)` do dustmasker -in $i -infmt fasta -outfmt fasta | sed -e '/>/!s/a\|c\|g\|t/N/g' > tempfile mv -f tempfile $i done
Some of the options available in Dustmasker:
Option Function -in <string> input file name -infmt <string> input format (e.g. fasta) -outfmt8 <string> output format (fasta)
Finally, we build the database with
kraken-build. With this command, Kraken uses all the masked genomes contained in the library (bacteria and mtDNA RefSeq) to create a database of 31 bp-long k-mers. We can choose the size of our custom database (hence the number of k-mers included, and the sensitivity) with the--max-db-sizeoption (8 Gb here).kraken-build --build --max-db-size 8 --db CustomDB
3.1.6. Taxonomic assignation with Kraken custom database¶
Once our custom database is built we can run the command for taxonomic assignation of DNA reads agaisnt the custom database, as in section 1.1 and 1.2.
kraken --db CustomDB --fastq-input merged.fastq.gz --gzip-compressed --output sample.kraken
kraken-report --db CustomDB sample.kraken > sample.kraken.report
Or, again, we can loop the commands if we have various samples.
for i in *.fastq
do
filename=$(basename "$i")
fname="${filename%.fastq}"
kraken --db CustomDB --threads 4 --fastq-input $i --output ${fname}.kraken
kraken-report --db CustomDB ${fname}.kraken > ${fname}.kraken.report
done
Finally, we can visualize the results of the Kraken analysis with Krona. Run ktImportTaxonomy to generate the html file and open it in a web browser to notice the difference with the analysis done with Minikraken.
ktImportTaxonomy -q 2 -t 3 sample.kraken -o sample.kraken.html
3.2. Kraken2¶
The first version of Kraken use a large indexed and sorted list of k-mer/LCA pairs as its database, which may be problematic for users due to the large memory requirements. For this reason Kraken 2 was developed. Kraken 2 is fast and requires less memory, BUT the database false positive errors occur in less than 1% of queries (confidence scoring thresholds can be used to call out to detect them). The default (or standard) database size is 29 GB (as of Jan. 2018, in Kraken 1 the standard database is about 200 GB!), and you will need slightly more than that in RAM if you want to build it. By default, Kraken 2 will attempt to use the dustmasker or segmasker programs provided as part of NCBI’s BLAST suite to mask low-complexity regions.
A Kraken 2 database is a directory containing at least 3 files:
- hash.k2d: Contains the minimizer to taxon mappings
- opts.k2d: Contains information about the options used to build the database
- taxo.k2d: Contains taxonomy information used to build the database
Other files may also be present as part of the database build process, and can, if desired, be removed after a successful build of the database.
3.2.1. Minikraken2¶
You can download the pre-built Minikraken2 database (containing bacteria, viral and archaea sequences) from the website with wget, and extract the archive content with tar:
wget ftp://ftp.ccb.jhu.edu/pub/data/kraken2_dbs/minikraken2_v1_8GB_201904_UPDATE.tgz
tar -xvzf minikraken2_v1_8GB_201904_UPDATE.tgz
To classify the reads in a fastq file against the Minikraken2 database, you can run this command:
kraken2 --db minikraken2_v1_8GB filename.fastq.gz --gzip-compressed --output filename.kraken --report filename.kraken.report
Some of the options available in Kraken 2:
| Option | Function |
|---|---|
| –use-names | Print scientific names instead of just taxids |
| –gzip-compressed | Input is gzip compressed |
| –report <string> | Print a report with aggregrate counts/clade to file |
| –threads | Number of threads (default: 1) |
Note
- In Kraken 2 you can generate the reports file by typing the
--reportoption (followed by a name for the report file to generate) in the command used for the classification. In Kraken 1, report files are generated with a specific command, after the classification (section 3.1.2: Create report files). - In order to run later Krona, the Kraken output file must contain taxids, and not scientific names. So if you want to run Krona do not call the option
--use-names.
The Kraken 2 report format, like Kraken 1, is tab-delimited with one line per taxon. There are six fields, from left to right:
- Percentage of fragments covered by the clade rooted at this taxon
- Number of fragments covered by the clade rooted at this taxon
- Number of fragments assigned directly to this taxon
- A rank code, indicating (U)nclassified, (R)oot, (D)omain, (K)ingdom, (P)hylum, (C)lass, (O)rder, (F)amily, (G)enus, or (S)pecies. Taxa that are not at any of these 10 ranks have a rank code that is formed by using the rank code of the closest ancestor rank with a number indicating the distance from that rank. E.g., “G2” is a rank code indicating a taxon is between genus and species and the grandparent taxon is at the genus rank.
- NCBI taxonomic ID number
- Indented scientific name
To visualize the results of the classification in multi-layerd pie charts, use Krona, as described in the section 3.1.3: Visualization of data with Krona
3.2.2. Kraken 2 Custom Database¶
We have already created a custom database to use in this hands-on session so we can go straight to the classification (step 4). However, we report here all the commands to build a Kraken2 database (steps 1-3).
The first step is to create a new folder that will contain your custom database (choose an appropriate name for the folder-database, here we will call it
CustomDB). Then we have to install a taxonomy. Usually, you will use the NCBI taxonomy. The following command will download in the folder/taxonomythe accession number to taxon maps, as well as the taxonomic name and tree information from NCBI:kraken2-build --download-taxonomy --db CustomDB
Install one or more reference libraries. Several sets of standard genomes (or proteins) are available, which are constantly updated (see also the Kraken website).
- archaea: RefSeq complete archaeal genomes/proteins
- bacteria: RefSeq complete bacterial genomes/proteins
- plasmid: RefSeq plasmid nucleotide/protein sequences
- viral: RefSeq complete viral genomes/proteins
- human: GRCh38 human genome/proteins
- fungi: RefSeq complete fungal genomes/proteins
- plant: RefSeq complete plant genomes/proteins
- protozoa: RefSeq complete protozoan genomes/proteins
- nr: NCBI non-redundant protein database
- nt: NCBI non-redundant nucleotide database
- env_nr: NCBI non-redundant protein database with sequences from large environmental sequencing projects
- env_nt: NCBI non-redundant nucleotide database with sequences from large environmental sequencing projects
- UniVec: NCBI-supplied database of vector, adapter, linker, and primer sequences that may be contaminating sequencing projects and/or assemblies
- UniVec_Core: A subset of UniVec chosen to minimize false positive hits to the vector database
You can select as many libraries as you want and run the following command, which will download the reference sequences in the folder
/library, as follows:kraken2-build --download-library bacteria --db CustomDB kraken2-build --download-library viral --db CustomDB kraken2-build --download-library plasmid --db CustomDB kraken2-build --download-library fungi --db CustomDB
In a custom database, you can add as many
fastasequences as you like. For exmaple, you can download the organelle genomes infastafiles from the RefSeq website with the commandswget:wget ftp://ftp.ncbi.nlm.nih.gov/refseq/release/mitochondrion/mitochondrion.1.1.genomic.fna.gz wget ftp://ftp.ncbi.nlm.nih.gov/refseq/release/mitochondrion/mitochondrion.2.1.genomic.fna.gz
The downloaded files are in compressed in the
gzformat. To unzip them run thegunzipcommand:gunzip mitochondrion.1.1.genomic.fna.gz gunzip mitochondrion.2.1.genomic.fna.gz
Then you can add the
fastafiles to your library, as follows:kraken2-build --add-to-library mitochondrion.1.1.genomic.fna --db CustomDB kraken2-build --add-to-library mitochondrion.2.1.genomic.fna --db CustomDB
Once your library is finalized, you need to build the database (here we set a maximum database size of 8 GB (you must indicate it in bytes!) with the
--max-db-sizeoption).kraken2-build --build --max-db-size 8000000000 --db CustomDB
Warning
Kraken 2 uses two programs to perform low-complexity sequence masking, both available from NCBI:
dustmasker, for nucleotide sequences, andsegmasker, for amino acid sequences. These programs are available as part of the NCBI BLAST+ suite. If these programs are not installed on the local system and in the user’s PATH when trying to usekraken2-build, the database build will fail. Users who do not wish to install these programs can use the--no-maskingoption to kraken2-build in conjunction with any of the--download-library,--add-to-library, or--standardoptions; use of the--no-maskingoption will skip masking of low-complexity sequences during the build of the Kraken 2 database.The
kraken2-inspectscript allows users to gain information about the content of a Kraken 2 database. You can pipe the command tohead, orless.kraken2-inspect --db path/to/dbfolder | head -5
Finally, we can run the classification of the reads against the custom database with the
kraken2command:kraken2 --db CustomDB filename.fastq.gz --gzip-compressed --output filename.kraken --report filename.report
To visualize the results of the classification in multi-layerd pie charts, use
Krona, as described in the section 3.1.3: Visualization of data with Krona
Note
Recently, a novel metagenomics classifier, KrakenUniq, has been developed to reduce false-positive identifications in metagenomic classification. KrakenUniq combines the fast k-mer-based classification of Kraken with an efficient algorithm for assessing the coverage of unique k-mers found in each species in a dataset. On various test datasets, KrakenUniq gives better recall and precision than other methods and effectively classifies and distinguishes pathogens with low abundance from false positives in infectious disease samples.
3.3. Mataphlan 3¶
MetaPhlAn is a computational tool that relies on ~1.1M unique clade-specific marker genes identified from ~100,000 reference genomes (~99,500 bacterial and archaeal and ~500 eukaryotic) to conduct taxonomic profiling of microbial communities (Bacteria, Archaea and Eukaryotes) from metagenomic shotgun sequencing data. MetaPhlAn allows:
- unambiguous taxonomic assignments;
- an accurate estimation of organismal relative abundance;
- species-level resolution for bacteria, archaea, eukaryotes, and viruses;
- strain identification and tracking
- orders of magnitude speedups compared to existing methods.
- metagenomic strain-level population genomics
To basic usage of MetaPhlAn for taxonomic profiling is:
metaphlan filename.fastq(.gz) --input_type fastq -o outfile.txt
Note
MetaPhlAn relies on BowTie2 to map reads against marker genes. To save the intermediate BowTie2 output use --bowtie2out, and for multiple CPUs (if available) use --nproc:
metaphlan filename.fastq(.gz) --bowtie2out filename.bowtie2.bz2 --nproc 5 --input_type fastq -o output.txt
The intermediate BowTie2 output files can be used to run MetaPhlAn quickly by specifying the input (--input_type bowtie2out):
metaphlan filename.bowtie2.bz2 --nproc 5 --input_type bowtie2out -o output.txt
For more information and advanced usage of MetaPhlAn see the manual and the wiki page (available for MetaPhlAn 2 at the moment).